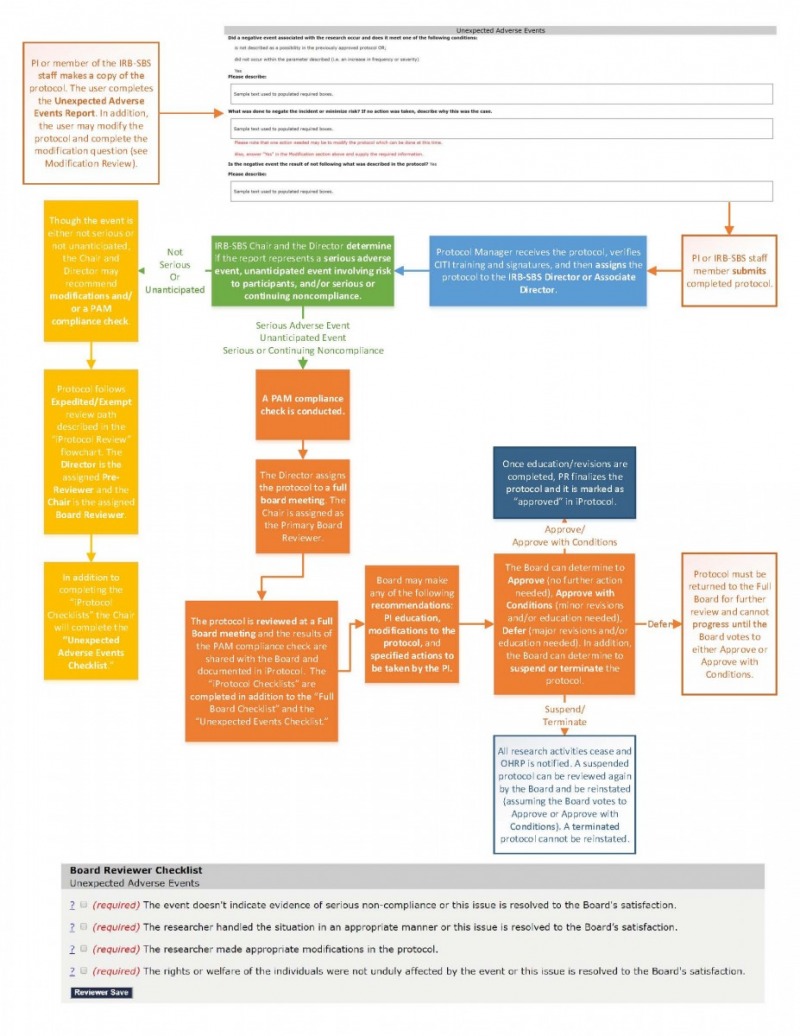
Research is not always predictable; in fact, some of the best discoveries have happened when things didn't go according to plan. However, there are instances in which you need to report unexpected events, particularly if the results are adverse. The purpose of reporting unexpected adverse events is not to be punitive but rather to provide an opportunity for the principal investigator to revise the protocol to prevent further issues and for the Board to understand where they can better assist researchers in the future.
Defining Unexpected Adverse Events
The following three categories define undesirable research events. These definitions are important to understanding what needs to be reported to the IRB-SBS and the Office for Human Research Protections (OHRP).
Unexpected Event
An unexpected event is an event that is not listed as a risk in the protocol and/or consent forms or was not previously documented at the intensity/frequency observed in your study population. The unexpected event also places the participant at greater risk than was previously known or recognized. For example, as a researcher in a typical school, you are interviewing children about a math test. During one of the interviews, the student tells you that his dad abuses him when he does poorly in school. Abuse is not a topic that you intended to cover in your study and the possibility of talking to students about abuse was not listed as a risk in your protocol. However, you are obligated to report child abuse and this is an unexpected event that changes the level of risk to the participant.
Adverse Event
An adverse event is any “undesirable and unintended, although not necessarily unexpected, effect of the research occurring in human subjects as a result of (a) the interventions and interactions used in the research; or (b) the collection of identifiable private information under the research” (Adapted from the 1993 OPRR IRB Guidebook). In other words, a participant is harmed as a result of participating in the study but the harm was described as a possibility in the protocol. For example, your study involves clinically depressed participants with a high suicidal rating. After attending intervention therapy as part of the study, one of the participants later in the evening reports a suicide attempt. This event was described as a risk in the protocol and in the consent form.
Serious Adverse Event
The potential harm for most studies fits in a scale of severity as related to probability. For example, the risks for a study measuring one’s happiness after exercising (low-impact moderate jogging) might include fatigue and soreness. Depending on the athleticism of the participants, the probability may be high for this adverse event to occur, though for normal, healthy adults this would not be considered a severe adverse event. A serious adverse event would be an event where the probability is not high for the event to occur and it resulted in harm to the participant. Continuing with the exercise example, one of the participants trips while performing the exercise and sprains an ankle, resulting in the need for medical assistance. Although the event is serious, it is still within the defined level of risk outlined in the protocol. However, if five of the fifteen participants receive the same injury, the frequency of the event changes the probability predicted in the protocol. The protocol should be reviewed to determine if the procedures need to be altered to better protect participants (i.e. select another jogging route) and if participants need to be better informed about the study (i.e. increased risk for injury, advice for safe jogging).
Adverse Event Related or Possibly Related to the study
It is easy to develop examples where an adverse event is clearly related to a study. However, it is not always clear that an injury or harm is directly related to a study and may require your best assessment of the situation. If you have questions about a potential situation, please contact our office. We would be happy to provide consultation.
Noncompliance Event
Noncompliance and more specifically, a "protocol violation," results from any change or departure from the study design or procedures of a research project that is not approved by the IRB-SBS prior to its implementation. Also, a protocol violation is thought to be any departure from established code of ethical conduct from the researcher's professional organization, or from applicable local, state, national and/or international regulations, policies and procedures. For example, a data recording procedure is not followed properly and confidential data about a participant is accidentally published on a website, or a laptop is stolen with participants’ social security numbers and home addresses, putting the participants at risk for identity theft. The act may be an unexpected problem or misfortune, a careless act or mistake, or a deliberate act against what the Board recommended.
In addition, failure to properly maintain a protocol is also considered a noncompliance event. For example, letting a protocol lapse without submitting a continuation request or modifying a protocol and failing to notify our office of the change are considered noncompliance events. Repeated disregard for maintaining a protocol is a serious noncompliance event. It is important that you keep accurate records and an updated protocol in order to maintain compliance with the IRB-SBS and federal regulations. If the study is complete, don't let it lapse but close the study instead.
When do I submit an Unexpected Adverse Events Report?
OHRP recognizes that adverse events are a normal part of conducting a study and while they should be recorded and monitored as part of the data collection process, they don’t necessarily need to be reviewed by the IRB and OHRP. OHRP does define three categories where reporting an adverse event is necessary:
- Adverse events that are serious, unexpected, and related or possibly related to participation in the research.
- Serious adverse events that are expected in some subjects, but are determined to be occurring at a significantly higher frequency or severity than expected.
- Other unexpected adverse events, regardless of severity, that may alter the IRB’s analysis of the risk versus potential benefit of the research and, as a result, warrant consideration of substantive changes in the research protocol or informed consent process/document.
Essentially, if the resulting event changes the level of risk that was previously approved by the Board, the Board and researcher needs to revisit the protocol’s procedures and recommend appropriate changes to accommodate the new level of risk. These changes could include changes to the protocol, consent forms, and other documents used in the study. If the protocol received an expedited review or was exempted in the initial review (because the protocol was considered minimal risk at the time), a change in risk may require that the protocol receive full board review for any future reviews. In addition, the Board has the authority to terminate or suspend approval for a study if they feel that the risk to participants is too great for the study to continue.
Noncompliance events that change the probability of harm to a participant must be reported to the IRB, even if a participant was not harmed as a result of the problem.
- Locate the protocol associated with the unexpected adverse event in the iProtocol Protocol Management page. Select the "create a copy" link below the protocol's title.
- Select the first radio button "I am copying in order to submit a Modification..."
- You will see a “Protocol was successfully updated” message. Click on the “return to protocol management” link to return to the Protocol Management page to find the new copy of the protocol.
- Select "edit" below the new copy of the protocol.
- Review the protocol. Please note that you can edit the protocol if needed but make sure to mark "yes" to the "Modifications" question below the "Continuation" question.
- In the "Unexpected Adverse Event" section near the end, complete the questions regarding the unexpected adverse event.
- When you are done making edits, select "submit" at the bottom of the page. In order for the submit button to be revealed, all of the "required" fields must be complete.
Trying to make a copy of the protocol for an unexpected events report but iProtocol won't allow it? iProtocol only allows one "in development" copy of the protocol for the same protocol number. Look for the protocol copy in the Protocol Management page. If you don't see it in your Protocol Management page, check the Trash/Recycle Bin for a copy of the protocol. Once you locate the modification copy, you have two options:
- Revise the copy that you already made for your current unexpected events report needs and submit it.
- Move the copy to the "Trash/Recycle Bin" and select the "permanent delete" button to delete the version (see Trash/Recycle Bin for directions). Once the copy is deleted, you can create a new copy from the most recently approved version of the protocol.
If you are completing the protocol for the first time and you do not need to report an unexpected adverse event, you should mark “no” to the first question and continue with the rest of the protocol. If you need to report an unexpected adverse event, mark "yes."
Did a negative event associated with the research occur and does it meet one of the following conditions:
- is not described as a possibility in the previously approved protocol OR
- did not occur within the parameter described (i.e. an increase in frequency or severity)
In this section, simply describe what occurred and provide details.
What was done to negate the incident or minimize risk? If no action was taken, describe why this was the case.
Provide the Board with any additional information that will help them understand the unexpected adverse event better. If you know what changes need to be made to negate or minimize risk going forward, it may be appropriate to modify the protocol. If this is the case, select “yes” to the modification questions and complete that section as well. You are not required to make a modification and it may not be appropriate to do so for every unexpected adverse event case.
Is the negative event the result of not following what was described in the protocol?
Including straightforward responses will help the Board understand the best course of action for the protocol. If the protocol wasn’t followed and the deviation resulted in the unexpected adverse event, following the protocol is a likely first solution. Part of the issue may be that the protocol isn’t practical or that there are issues with the original plan that need to be addressed so that it can be followed. Again, it may be appropriate to proactively modify the protocol in addition to completing this section and the two can be done together.